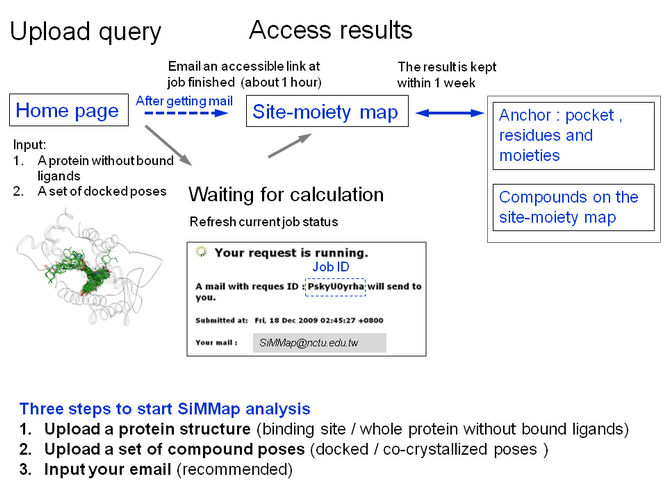
| What can SiMMap analysis do for you? |
SiMMap server provides analysis of Site-Moiety Map. The SiMMap server statistically derives site-moiety map with several anchors,
which describe the relationship between the moiety preferences and physico-chemical properties of the binding site, from the interaction profiles between
query target protein and its docked (or co-crystallized) compounds. Each anchor includes three basic elements: a binding pocket with conserved interacting
residues, the moiety composition of query compounds, and pocket-moiety interaction type (electrostatic, hydrogen-bonding, or van der Waals).
To start the SiMMap analysis, you need to upload a protein structure (X-ray structure,
or single model of NMR/simulated structure) and a set of compounds (docked
poses or aligned co-crystallized poses). Our server will automatically generate site-moiety map and statistics, and then visualize them on the result pages.
Please note that SiMMap does not provide docking services
|
| How to use SiMMap server |
Acceptable protein structure : X-ray structure, or single model of NMR/simulated structure
A set of compounds : Docked poses or aligned co-crystallized poses
If you have any problems or suggestions for our server, please let us know (contact us).
|
| Examples |
The example cases with target proteins and virtual screening compound sets are shown as below. Each compound set consists of known active ligands and 990 randomly chosen
non-active compounds from the ACD
Antiagonist forms of estrogen receptor α (EST, PDB id : 3ert): estrogen receptor alpha (ERα),
proposed by Bissantz et al. to evaluate the screening utility of virtual screening capability
Thymidine kinase (TK, PDB id: 1kim): The test set for virtual screening against HSV-1 thymidine
kinase (TK) was proposed by Bissantz et al. (Journal of Medicinal Chemistry, 43:4759-4767, 2000), including 10 known active ligands of TK and 990 randomly chosen
non-active compounds from the ACD.
|
| Upload a target protein by user (protein structure) |
Acceptable protein structure : X-ray structure, or single model of NMR/simulated structure
To save the calculation time, we suggest users to upload the structure of binding site.
The uploaded protein structure can be whole protein or a part of the protein (e.g., binding site). The uploaded structure should not contain any bound ligands
(except for cofactors, key structure waters or modified amino acids).
Acceptable format of protein structure is PDB format (.pdb file). The protein structure file needs to be ".pdb" and contain
3D coordinates.
The HETATM records in the binding site are default to remove. The records present
the atomic coordinate records for atoms within "non-standard" groups in PDB file. If you need to retain HETATM records (e.g., cofactors , key structure waters or modified
amino acids) in the upload protein structure as a part of a protein, please select retain HETATM records in protein.
|
| Upload a compressed file for a set of docked compounds (molecular formats) |
A set of docked compounds : Docked poses or aligned co-crystallized poses. Please note that SiMMap does not
provide docking services
We suggest to use mol/mol2 formats for the uploaded compounds. Each PDB ligand will require a brief time for checking correctness (HETATM and CONECT blocks).
If users need to convert PDB into mol/mol2, some public tools (e.g., OpenBabel and
JOELib) are freely available. (How to convert by openbable)
For reducing uploading time, a compressed file of docked compounds is required. The acceptable formats of compressed files are ZIP and TAR (.zip and .tar).
Currently, the maximum number of compounds is 3000 for online service.
Examples for acceptable compound formats: 2’-deoxythymidine (mol, mol2 and pdb)
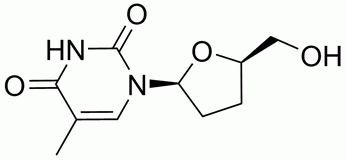
- MDL mol
An acceptable Mol file consists of a header block and a connection table. The format document is described on symyx web site.
- SYBYL mol2
A Mol2 file (.mol2) is a complete, portable representation of a SYBYL molecule. The format document is described on Tripos web site.
- PDB
An acceptable PDB format needs to consists of an atom block (HETATM), a connection table (CONECT). The atom block and connection table are as same as PDB document.
|
| How to compress your molecule files as a .zip or .tar file |
Please save compound files in the ".mol2", ".mol" or ".pdb" formats and then compress these compounds into a ".zip" or ".tar" file.
Zip files may be created using a utility such as JZip.
For example, a set of docked compounds in the folder, "MyCompounds" to compress as the file, "compounds"
Following steps will help you generate this zip/tar file for uploading.
For zip file : on windows, DOS or linux
> zip molecules.zip .\MyCompounds
Or Right click target directory. Select the term of the menu, "Send to" -> "zipped directory"
For tar file : On linux or Mac
>tar -czf molecules.tar ./MyCompounds
|
| e-mail notification |
This server takes a brief time (up to several hours) to process analysis. The calculation time mainly depends on the number of query compounds.
The server will automatically send a notification mail with the result link to users. The browser will be re-directed to results when the job is completed.
|
| SiMMap: site-moiety map (anchors) |
The SiMMap server statistically derives site-moiety map with several anchors, which describe the relationship between the moiety preferences and physico-chemical properties
of the binding site, from the interaction profiles between query target protein and its docked (or co-crystallized) compounds.
Each anchor includes three basic elements:
1. A binding pocket with conserved interacting residues
2. The moiety composition of query compounds
3. Pocket-moiety interaction type (electrostatic, hydrogen-bonding, or van der Waals)
An anchor is often a hot spot and the site-moiety map can help to assemble potential leads by visualizing the interactions of optimal steric, hydrogen-bonding, and
electronic moieties for binding pockets. When a compound highly agrees with anchors of site-moiety map, this compound often activates or inhibits the query target.
We believe that the site-moiety map is useful for drug discovery and understanding biological mechanisms.
For example, P-loop (Walker A motif) of nucleotide-binding proteins is a glycine-rich loop and ATP/GTP binding motif.
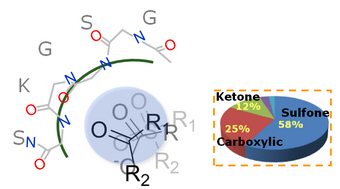
An anchor for P-loop and related compound moieties in SiMMap
Consensus interactions of interacting residues and moieties (functional groups of docked compounds) was identified in SiMMap. For example, the phosphate, sulfate, and
carboxyl groups can be often identified and interacted with residue backbones of P-loop in post-screening analysis.
|
| Score of SiMMap |
One application of SiMMap is to identify active compounds (substrates or inhibitors) of query protein. Generally, a compound often activates or inhibits the query target when
its interactions and moieties highly agree with anchors of site-moiety map.
SiMMap scores a compound by combining predicted binding energy of GEMDOCK and the anchor scores between the map and the compound. The score, S(x) is defined as
S(x) = Σ(n, i=1) ASi(x) + wEGEMDOCK
where the ASi(x) is 1 if the moiety of the compound x agrees the anchor i on the target protein.
n is number of anchors. EGEMDOCK is the predicted binding energy of GEMDOCK.
w is the weight. Based on SiMMap scores, we can obtain new ranks of query compounds.
|
| Energy between interacting residue and moiety |
The energy (unit: kcal/mol) between interacting residue and moiety is calculated by GEMDOCK piecewise linear potential (PLP). GEMDOCK (Generic Evolutionary Method for molecular
DOCKing) is free for non-commercial researches. It is a docking tool for computing a ligand conformation and orientation relative to the active site of target protein.
|
| z-score (identification of consensus interactions) |
We used z-score value to measure the interacting conservation between this residue and moieties. The standard deviation (σ) and mean (μ) were derived by random shuffling
1,000 times in interacting profiles. The interactions of each protein-compound are considered as a binomial distribution. With a set of compounds, the binomial distribution
is capable to approach normal distribution. According to the profiles, we infer anchor candidates by identifying the pockets with significant interacting residues and
moieties with z-score >= 1.645.
|
| 3D presentation of site-moiety map |
The result will be shown as data tables and visualized in 3D presentation. Jmol applet is embedded for 3D presentation. We suggest users' browser to install
Java Runtime Environment for running Jmol applet.
|
| Presentation of compound structures |
We present compound structures by using OASA library which is a python library for manipulation of chemical formats that forms the base of BKChem.
OASA library of BKChem is a free chemical drawing program. It was conceived and written by Beda Kosata. In some cases, the figures cannot be correctly generated (ex: bulky circles or closed substitution groups). These problems will not interfere with our analysis.
|
